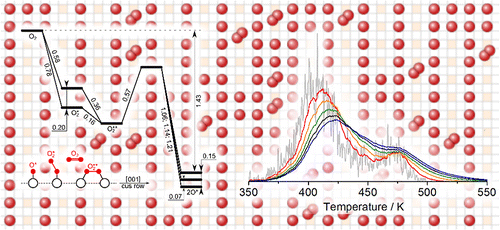
The theoretical study of catalysis would substantialy benefit from the use of atomistic simulations that can provide information beyond mean-field approaches. To date, the nanoscale understanding of surface reactions has been only qualitatively achieved by means of kinetic Monte Carlo coupled to density functional theory, KMC-DFT. Here, we examine a widely employed model for oxygen interaction with the RuO2(110) surface, a highly anisotropic system. Our analysis reveals several covert problems that render as questionable the model’s predictions. We suggest an advanced approach that considers all the relevant elementary steps and configurations while smoothing the intrinsic errors in the DFT description of oxygen. Under these conditions, KMC provides quantitative agreement to temperature-programmed desorption experiments. These results illustrate how KMC-based simulations can be pushed forward so that they evolve toward being the standard methodology to study complex chemistry at the nanoscale.