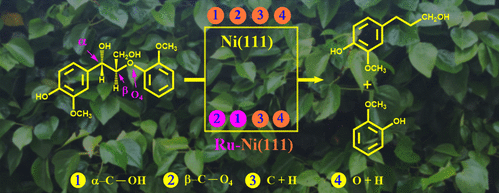
Efficient lignin depolymerization is crucial for achieving a positive economical return in biorefineries. However, because of the complex cross-polymerized network formed by the aromatic lignols, catalysts that can work at low temperatures perform insufficiently in terms of both activity and selectivity. Recently, Ni-based catalysts have been reported to exhibit a superior catalytic behavior in lignin valorization, but the mechanism of the depolymerization remains unclear. In this study, we employed density functional theory to investigate lignin decomposition on pure and Ru-doped Ni(111) surfaces to unravel the key issues that limit performance. The reaction network was screened by using complex coniferyl dimer models containing different stereocenters to simulate the most abundant β-O-4 linkages in lignin, thus presenting a better representation of the complexity of the parent compound. Our results show that on nickel, both adsorption geometries and the preferred reaction paths are different depending on the chirality of the dimer reactants. In addition, the rigidity of the β-O-4 link is also responsible for the reactivity found. Finally, the presence of small amounts of Ru on the surface simplifies the reaction as they act as preferential points for β-O-4 bond cleavage via the weakened C–O bond that is induced by increasing metal–O bond strength.