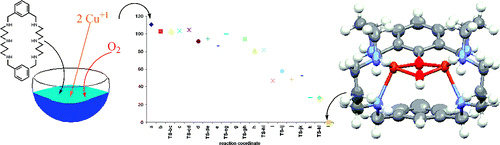
The present study reports the first example of a complete and detailed mechanism of intramolecular aromatic hydroxylation through O2 activation by a hexaazamacrocyclic dicoppper(I) complex, [CuI2(H3m)]2+. The reactivity of this complex has been previously studied experimentally, although only the characterization of the final μ-phenoxo-μ-hydroxo [CuII2(H3m-O)(μ-OH)]2+ product was possible. In the present theoretical study, we unravel the reaction pathway for the overall intramolecular aromatic hydroxylation, that is, from the initial reaction of O2 with the dicopper(I) species to the final [CuII2(H3m-O)(μ-OH)]2+ product using the B3LYP method. Our results indicate that a CuICuII-superoxo species is formed first, then the interaction of the O2 moiety with the second CuI center leads to a μ-η2:η2-peroxo-CuII2 intermediate. This latter species is found to be close in energy with the isomeric bis(μ-oxo) species. The relative stability of these two isomers depends on the method of calculation, and therefore, it has not been possible to reach a definite conclusion about the nature of the active species in this reaction mechanism. Notwithstanding, our B3LYP calculations indicate that it is the μ-η2:η2-peroxo species that evolves via an electrophilic σ* attack involving a concerted peroxide O-O bond cleavage and C-O bond formation to a Wheland-type intermediate. The reaction follows with a proton release assisted by the presence of a second aromatic ring yielding a μ-hydroxo-μ-oxo intermediate species, which, in the final stage of the reaction, rearranges to the product. The proton transfer path points out the possibility to design new systems with improved reactivity by properly placing a second aromatic ring to assist the deprotonation step. The lack of high energy barriers and deep energy wells explains the difficulty to trap intermediates experimentally.