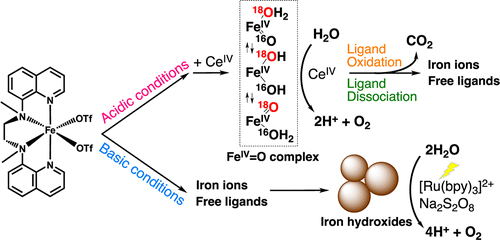
Thermal water oxidation by cerium(IV) ammonium nitrate (CAN) was catalyzed by nonheme iron complexes, such as Fe(BQEN)(OTf)2 (1) and Fe(BQCN)(OTf)2 (2) (BQEN = N,N′-dimethyl-N,N′-bis(8-quinolyl)ethane-1,2-diamine, BQCN = N,N′-dimethyl-N,N′-bis(8-quinolyl)cyclohexanediamine, OTf = CF3SO3–) in a nonbuffered aqueous solution; turnover numbers of 80 ± 10 and 20 ± 5 were obtained in the O2 evolution reaction by 1 and 2, respectively. The ligand dissociation of the iron complexes was observed under acidic conditions, and the dissociated ligands were oxidized by CAN to yield CO2. We also observed that 1 was converted to an iron(IV)-oxo complex during the water oxidation in competition with the ligand oxidation. In addition, oxygen exchange between the iron(IV)-oxo complex and H218O was found to occur at a much faster rate than the oxygen evolution. These results indicate that the iron complexes act as the true homogeneous catalyst for water oxidation by CAN at low pHs. In contrast, light-driven water oxidation using [Ru(bpy)3]2+ (bpy = 2,2′-bipyridine) as a photosensitizer and S2O82– as a sacrificial electron acceptor was catalyzed by iron hydroxide nanoparticles derived from the iron complexes under basic conditions as the result of the ligand dissociation. In a buffer solution (initial pH 9.0) formation of the iron hydroxide nanoparticles with a size of around 100 nm at the end of the reaction was monitored by dynamic light scattering (DLS) in situ and characterized by X-ray photoelectron spectra (XPS) and transmission electron microscope (TEM) measurements. We thus conclude that the water oxidation by CAN was catalyzed by short-lived homogeneous iron complexes under acidic conditions, whereas iron hydroxide nanoparticles derived from iron complexes act as a heterogeneous catalyst in the light-driven water oxidation reaction under basic conditions.