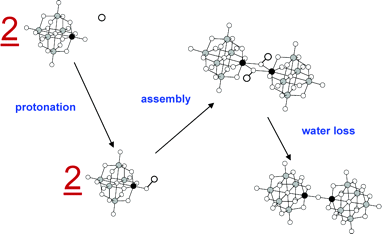
Density functional theory (DFT) calculations are used to investigate dimerization, by acid condensation, between Lindvist and Keggin cluster anions. When specific addendum atoms are present (notably, Ti, V, Nb, and Cr), these clusters dimerize in the presence of acid via the formation of nu-O linkages. These processes may be viewed as molecular models for the formation of metal-oxygen bonds in the active sites of metalloenzymes, or in materials chemistry, and we here provide detailed information concerning these reactions. For this, DFT calculations are used to provide insight into the mechanism(s) associated with dimerization of Lindqvist clusters, W5MO19n- (M = W, Mo, V, Nb, and Ti) in acidic media. In full accord with experimental data, our calculations show that dimerization of Nb and Ti derivatives is thermodynamically favored and that this is not the case for dimerization between Mo, V, and W addendum atoms. In addition, dimerization of PW11TiO405- and SiW11NbO405- to give the corresponding dimers was also calculated to be exothermic. Two possible mechanisms, involved in two competing pathways, are evaluated and discussed. Conclusions are presented concerning the role played by the nature of the metal atoms taking part in M–O-M bond formation. A correlation is established between the relative strengths of specific terminal M=O bonds in monomeric precursors and the tendency of these to form M–O-M bonds.