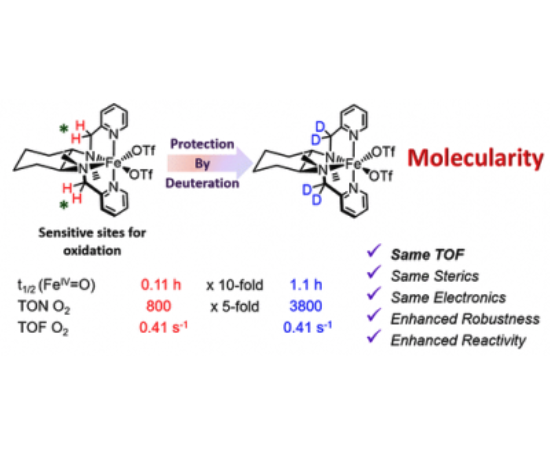
The nature of the oxidizing species in water oxidation reactions with chemical oxidants catalyzed by α-[Fe(OTf)2(mcp)] (1α; mcp = N,N′-dimethyl-N,N′-bis(pyridin-2-ylmethyl)cyclohexane-1,2-diamine, OTf = trifluoromethanesulfonate anion) and β-[Fe(OTf)2(mcp)] (1β) has been investigated. Mössbauer spectroscopy provides definitive evidence that 1α and 1β generate oxoiron(IV) species as the resting state. Decomposition paths of the catalysts have been investigated by identifying and quantifying ligand fragments that form upon degradation. This analysis correlates the water oxidation activity of 1α and 1β with stability against oxidative damage of the ligand via aliphatic C–H oxidation. The site of degradation and the relative stability against oxidative degradation are shown to be dependent on the topology of the catalyst. Furthermore, the mechanisms of catalyst degradation have been rationalized by computational analyses, which also explain why the topology of the catalyst enforces different oxidation-sensitive sites. This information has served in creating catalysts where sensitive C–H bonds have been replaced by C–D bonds. The deuterated analogues D4-α-[Fe(OTf)2(mcp)] (D4-1α), D4-β-[Fe(OTf)2(mcp)] (D4-1β), and D6-β-[Fe(OTf)2(mcp)] (D6-1β) were prepared, and their catalytic activity has been studied. D4-1α proves to be an extraordinarily active and efficient catalyst (up to 91% of O2 yield); it exhibits initial reaction rates identical with those of its protio analogue, but it is substantially more robust toward oxidative degradation and yields more than 3400 TON (n(O2)/n(Fe)). Altogether this evidences that the water oxidation catalytic activity is performed by a well-defined coordination complex and not by iron oxides formed after oxidative degradation of the ligands.