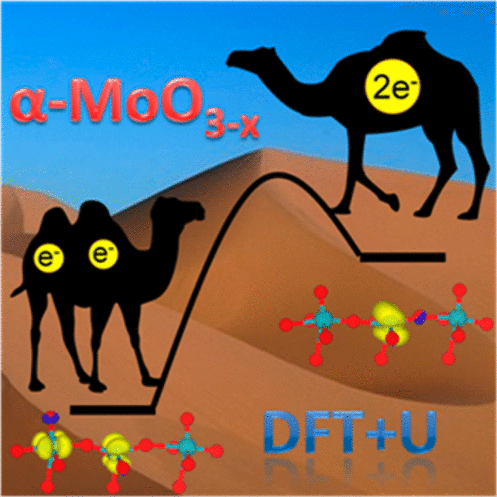
Molybdenum oxides are finding increasing applications that rely on their redox character. For the most common polymorph, α-MoO3, oxygen vacancy formation leaves two electrons on the surface that can be stored as small polarons. Detailed density functional theory calculations that properly account for the self-interaction term, Ueff = 3.5 eV, show that the vacancy generates two different configurations: either two Mo5+ centers (Mo5+□ and Mo5+═O) or a single double-reduced Mo4+. These states are separated by 0.22 eV with a barrier for interconversion of 0.33 eV, and thus both are populated at catalytic temperatures, as shown by first-principles molecular dynamics. At higher reduction levels, vacancies can only be accumulated along a preferential direction and the energy difference between the 2×Mo5+ and Mo4+ configurations is reduced. These results point out the need for a revision of the experimental assignments based on our characterization that includes charges, vibrational frequencies, and XPS signatures.