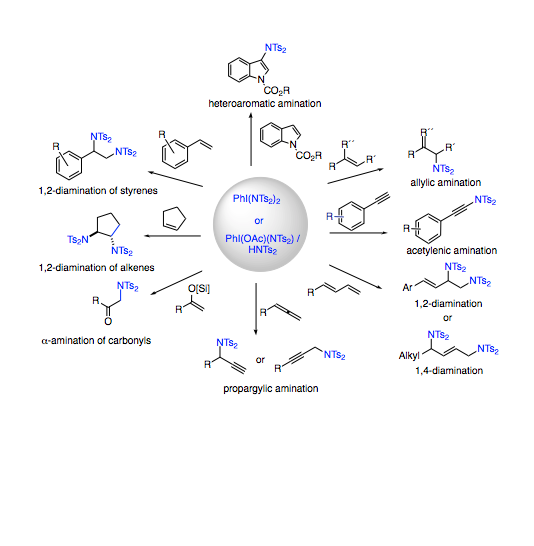
The quest for the development of new protocols that provide general conditions for oxidative carbon-nitrogen bond formation has grown over recent years. Within this context, due to feasibility and benignity considerations in biochemical sciences, reactions that rely on main group oxidants as the only promoters have received particular interest. We have recently found that simple protonolysis events enable the incorporation of nitrogenated groups of the bissulfonimide family into the coordination sphere of common iodine(III) complexes such as diacetoxy iodobenzene. The products of the type ArI(OAc)(NTs2) represent rare examples of iodine(III) compounds displaying reactive iodine-nitrogen single bonds. Further protonolysis furnishes the corresponding iodine(III) compounds ArI(NTs2)2 containing two defined iodine-nitrogen single bonds for unprecedented dual transfer of both nitrogenated groups. It is of great synthetic importance that these new compounds contain iodine-nitrogen entities, which upon dissociation in solution lead to electrophilic iodine centers and nucleophilic nitrogen groups. This has enabled the development of a body of conceptually new amination reactions, which do not rely on conventional electrophilic nitrogen reagents but rather employ iodine(III) as an electrophilic activator and bissulfonimides as the source of subsequent nucleophilic amination. Additional diversification arises from the ambident nature of bissulfonimines enabling oxygenation pathways. The exciting chemistry covered in this Account comprises structural features of the reagents (including X-ray analysis), scope and limitation in synthetic amination of different hydrocarbons (including sp-, sp2-, and sp3-hybridized centers as in acetylenes, alkenes, enols, butadienes, allenes, arenes, and alkylketones), and physical-organic and theoretical analysis of the underlying reaction mechanisms. The oxidative transformations with all their rich diversifications originate from the versatile redox chemistry of the iodine(III) and iodine(I) pair, which shares several aspects of transition metal high oxidation state chemistry. For the present aryliodine(III) reagents, steric and electronic fine-tuning is possible through accurate engineering of the arene substituent. In addition to the general reactivity of the I-N bond, chiral aryliodine(III) reagents with defined stereochemical information in the aryl backbone are conceptually compatible with this approach. Thus, the development of enantioselective amination reactions with up to 99% ee was also successful. Several of the active enantioselective reagents have been isolated and structurally characterized. Following this approach for the important class of chiral vicinal diamines, an unprecedented direct diamination of alkenes could be conducted in an enantioselective catalytic manner under full intermolecular reaction control. This latter reaction is based on the precise engineering of a chiral aryliodine(III) catalyst in combination with bismesylimide as nitrogen source. It is the consequence of the precise understanding of the reaction behavior of structurally defined bisimidoiodine(III) reagents.