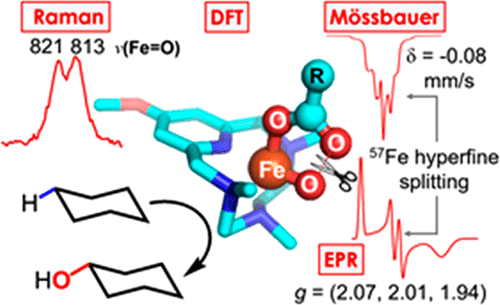
The reaction of [(PyNMe3)FeII(CF3SO3)2], 1, with excess peracetic acid at −40 °C generates a highly reactive intermediate, 2b(PAA), that has the fastest rate to date for oxidizing cyclohexane by a nonheme iron species. It exhibits an intense 490 nm chromophore associated with an S = 1/2 EPR signal having g-values at 2.07, 2.01, and 1.94. This species was shown to be in a fast equilibrium with a second S = 1/2 species, 2a(PAA), assigned to a low-spin acylperoxoiron(III) center. Unfortunately, contaminants accompanying the 2(PAA) samples prevented determination of the iron oxidation state by Mössbauer spectroscopy. Use of MeO-PyNMe3 (an electron-enriched version of PyNMe3) and cyclohexyl peroxycarboxylic acid as oxidant affords intermediate 3b(CPCA) with a Mössbauer isomer shift δ = −0.08 mm/s that indicates an iron(V) oxidation state. Analysis of the Mössbauer and EPR spectra, combined with DFT studies, demonstrates that the electronic ground state of 3b(CPCA) is best described as a quantum mechanical mixture of [(MeO-PyNMe3)FeV(O)(OC(O)R)]2+ (∼75%) with some FeIV(O)(•OC(O)R) and FeIII(OOC(O)R) character. DFT studies of 3b(CPCA) reveal that the unbound oxygen of the carboxylate ligand, O2, is only 2.04 Å away from the oxo group, O1, corresponding to a Wiberg bond order for the O1–O2 bond of 0.35. This unusual geometry facilitates reversible O1–O2 bond formation and cleavage and accounts for the high reactivity of the intermediate when compared to the rates of hydrogen atom transfer and oxygen atom transfer reactions of FeIII(OC(O)R) ferric acyl peroxides and FeIV(O) complexes. The interaction of O2 with O1 leads to a significant downshift of the Fe–O1 Raman frequency (815 cm–1) relative to the 903 cm–1 value predicted for the hypothetical [(MeO-PyNMe3)FeV(O)(NCMe)]3+ complex.