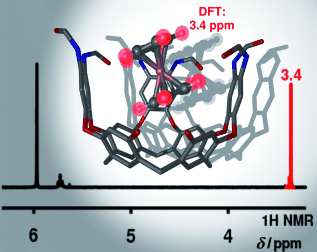
We describe herein a detailed study of the inclusion processes of several positively charged organometallic sandwich complexes inside the aromatic cavity of the self-folding octaamide cavitand 1. In all cases, the binding process produces aggregates with a simple 1:1 stoichiometry. The resulting inclusion complexes are not only thermodynamically stable, but also kinetically stable on the 1H NMR spectroscopy timescale. The binding constants for the inclusion complexes were determined by different titration techniques. We have also investigated the kinetics of the binding process and the motion of the metallocenes included in the aromatic cavity of the host. Using DFT-based calculations, we have evaluated the energies of a diverse range of potential binding geometries for the complexes. We then computed the proton chemical shifts of the included guest in each one of the binding geometries. The agreement between the averaged computed values and the experimentally determined chemical shifts clearly supports the proposed binding geometries that we assigned to the inclusion complexes formed in solution. The combination of experimental and theoretical results has allowed us to elucidate the origins of the distinct features detected in the complexation process of the different guests, as well as their different motions inside the host.