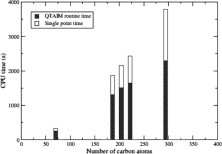
An improved version of our method for computing QTAIM [J. I. Rodríguez, A. M. Köster, P. W. Ayers, A. Santos-Valle, A. Vela, G. Merino, J. Comput. Chem. 2009, in press, doi:10.1002/jcc.21134] is presented. Vectorization and parallelization of the previous algorithm, together with molecular symmetry, make the present algorithm as much as two orders of magnitude faster than our original method. The present method scales linearly with both system size and the number of processors. The performance of the method is demonstrated by computing the QTAIM atomic properties of a series of carbon nanotubes. Our results show that the CPU time for a QTAIM property calculation is comparable to that of a SCF-single point calculation. The accuracy of the original method is also improved.