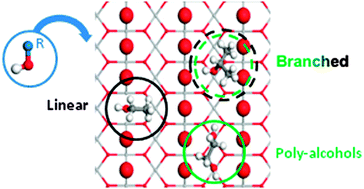
We have studied by means of density functional theory including dispersion contributions, the interaction of small chain alcohols with up to four carbons and three hydroxyl groups on the TiO2(110) rutile surface with different reduction degrees. Adsorption takes place through an acid–base interaction that can lead to both molecular and dissociated species. The latter are energetically preferred. Bulk reduction does not apport significant change neither in the structure nor in the adsorption energies, because the electrons are delocalized to a great extent. If vacancies are present at the surface these are the best adsorption sites for primary and secondary monoalcohols. Tertiary or poly-alcohols prefer the Ticus channels, but the reasons for the site preference are different. In the case of bulky alcohols, steric hindrance is the main adsorption-controlling factor, while templating effects of the basic (oxygen) sites on the surface are the key parameters to understand the adsorption of poly-alcohols. Vicinal polyalcohols behave even in a more complex way, for that they prefer the vacancy position only when dissociated, otherwise they stay in the Ticus channel. Our results warn about the use of small surrogates to investigate the chemistry of large alcohols as the adsorption patterns are not only quantitatively but also qualitatively wrong.