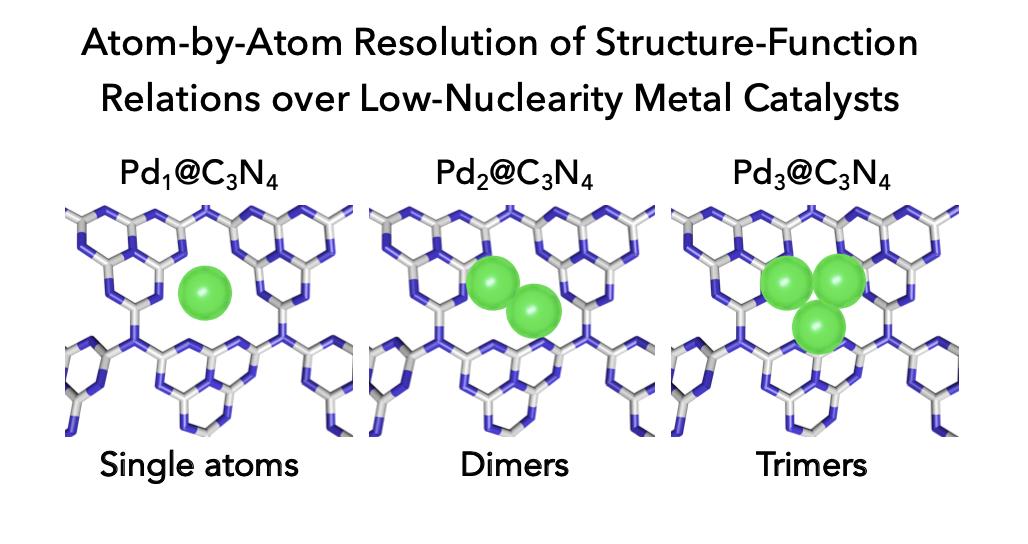
Controlling the structure sensitivity of catalyzed reactions over metals is central to developing atom‐efficient chemical processes. Approaching the minimum ensemble size, the properties enter a non‐scalable regime where each atom counts. Almost all trends in this ultra‐small frontier derive from surface science approaches using model systems, because of both synthetic and analytical challenges. Exploiting the unique coordination chemistry of carbon nitride, we discriminate through experiments and simulations the interplay between the geometry, electronic structure, and reactivity of palladium atoms, dimers, and trimers. Catalytic tests evidence application‐dependent requirements of the active ensemble. In the semi‐hydrogenation of alkynes, the nuclearity primarily impacts activity, whereas the selectivity and stability are affected in Suzuki coupling. This powerful approach will provide practical insights into the design of heterogeneous catalysts comprising well‐defined numbers of atoms.