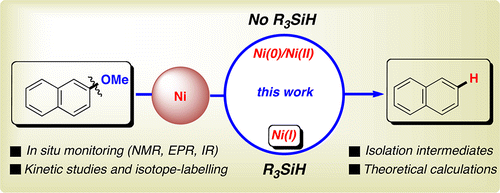
A mechanistic and computational study on the reductive cleavage of C-OMe bonds catalyzed by Ni(COD)2/PCy3 with silanes as reducing agents is reported herein. Specifically, we demonstrate that the mechanism for this transformation does not proceed via oxidative addition of the Ni(0) precatalyst into the C-OMe bond. In the absence of an external reducing agent, the in-situ-generated oxidative addition complexes rapidly undergo β-hydride elimination at room temperature, ultimately leading to either Ni(0)-carbonyl- or Ni(0)-aldehyde-bound complexes. Characterization of these complexes by X-ray crystallography unambiguously suggested a different mechanistic scenario when silanes are present in the reaction media. Isotopic-labeling experiments, kinetic isotope effects, and computational studies clearly reinforced this perception. Additionally, we also found that water has a deleterious effect by deactivating the Ni catalyst via formation of a new Ni-bridged hydroxo species that was characterized by X-ray crystallography. The order in each component was determined by plotting the initial rates of the C-OMe bond cleavage at varying concentrations. These data together with the in-situ-monitoring experiments by 1H NMR, EPR, IR spectroscopy, and theoretical calculations provided a mechanistic picture that involves Ni(I) as the key reaction intermediates, which are generated via comproportionation of initially formed Ni(II) species. This study strongly supports that a classical Ni(0)/Ni(II) for C-OMe bond cleavage is not operating, thus opening up new perspectives to be implemented in other related C-O bond-cleavage reactions.