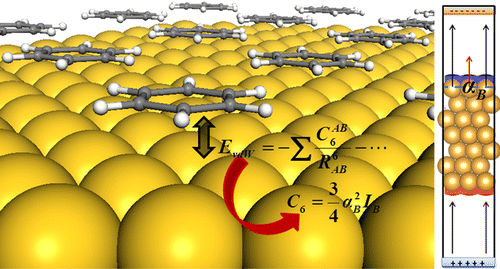
Many common density functional theory methods used in the study of adsorption on metals lack dispersion contributions. Formulations like the random phase approximations would mitigate this error, but they are computationally too expensive. Therefore, semiempiric treatments based on dispersion coefficients turn out to be a practical solution. However, the parameters derived for atoms and molecules are not easily transferable to solids. In the case of metals, they cause severe overbinding as screening is not properly taken into consideration. Alternative ways to determine these parameters for metal surfaces have been put forward, but they are complex and not very flexible when employed to address low-coordinated atoms or alloys. In this work, we present a self-consistent, fast, and costless tool to obtain the dispersion coefficients for metals and alloys for pristine and defective surfaces. Binding energies computed with these parameters are compared to both the experimental and theoretical values in the literature thus demonstrating the validity of our approach.