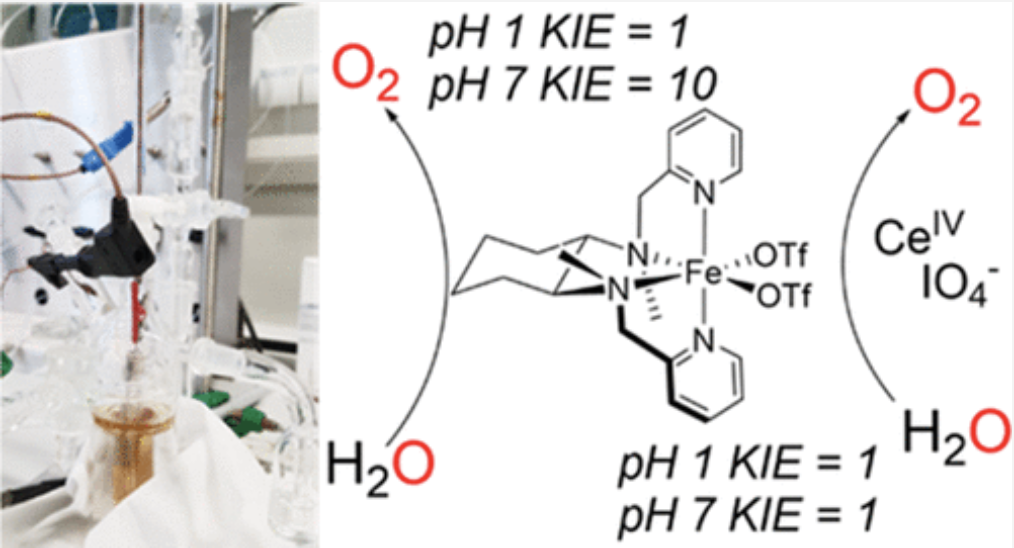
The complex α-[Fe(mcp)(OTf)2] (mcp = N,N′-dimethyl-N,N′-bis(pyridin-2-ylmethyl)-cyclohexane-1,2-diamine and OTf = trifluoromethanesulfonate anion) was reported in 2011 by some of us as an active water oxidation (WO) catalyst in the presence of sacrificial oxidants. However, because chemical oxidants are likely to take part in the reaction mechanism, mechanistic electrochemical studies are critical in establishing to what extent previous studies with sacrificial reagents have actually been meaningful. In this study, the complex α-[Fe(mcp)(OTf)2] and its analogues were investigated electrochemically under both acidic and neutral conditions. All the systems under investigation proved to be electrochemically active toward the WO reaction, with no major differences in activity despite the structural changes. Our findings show that WO-catalyzed by mcp–iron complexes proceeds via homogeneous species, whereas the analogous manganese complex forms a heterogeneous deposit on the electrode surface. Mechanistic studies show that the reaction proceeds with a different rate-determining step (rds) than what was previously proposed in the presence of chemical oxidants. Moreover, the different kinetic isotope effect (KIE) values obtained electrochemically at pH 7 (KIE ∼ 10) and at pH 1 (KIE = 1) show that the reaction conditions have a remarkable effect on the rds and on the mechanism. We suggest a proton-coupled electron transfer (PCET) as the rds under neutral conditions, whereas at pH 1 the rds is most likely an electron transfer (ET).