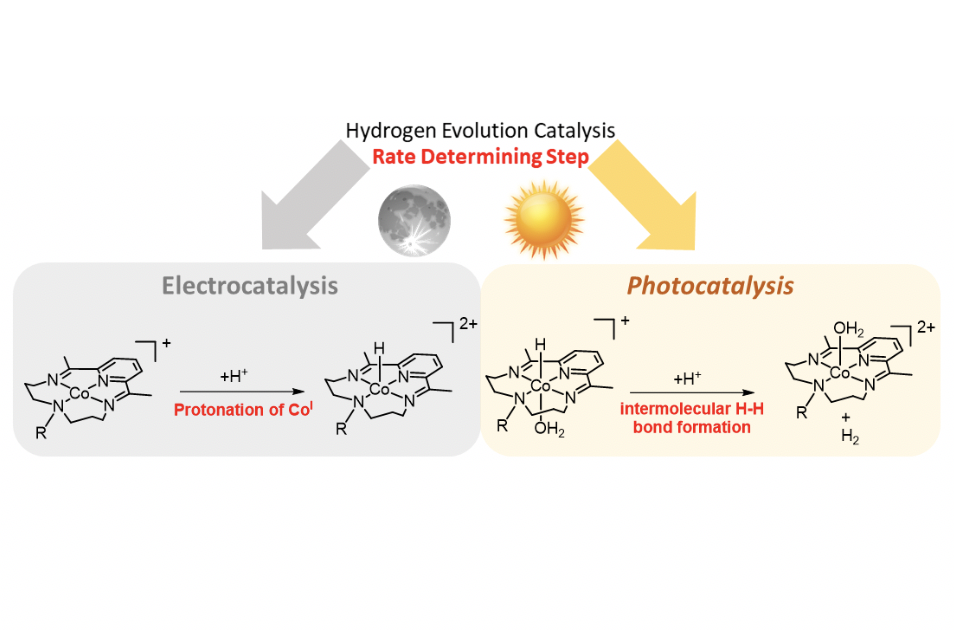
Cobalt complexes containing equatorial tetraazamacrocyclic ligands are active catalysts for the hydrogen evolution reaction in pure aqueous conditions. Herein we explore the effect of different groups directly linked to the macrocyclic ligand (‐NH‐, ‐NH2+‐ or –N(CH2OH)‐). Electrochemically induced hydrogen evolution catalysis studies at pH 4 invoke a mechanism, in which the rate determining step is the protonation of the reduced CoI species that gives a cobalt hydride (CoIII‐H), a key intermediate towards the H‐H bond formation. In sharp contrast, under photochemical conditions using [Ru(bpy)3]2+ as a photosensitizer and ascorbate as sacrificial electron donor, the formation of a “Co0” species that quickly protonates to give a CoII‐H is proposed. In this scenario, the rate determining step is the H‐H bond formation that occurs in an intermolecular fashion from the CoII‐H species and a water molecule. Both mechanisms are supported by density functional theory (DFT) calculations that allowed us to estimate the pKa values of the CoIII‐H and CoII‐H species, as well as transition states based on intramolecular and intermolecular H‐H bond formation from CoII‐H.