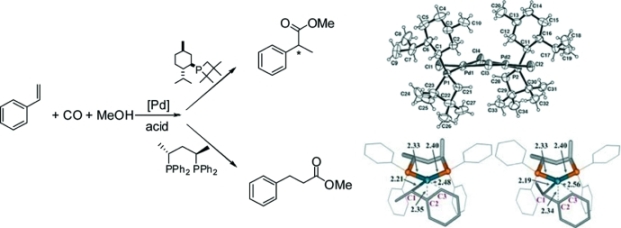
Catalytically active palladium systems bearing the monodentate phosphetane ligand 1 and diphosphane 2 were studied by multinuclear NMR and HP-NMR spectroscopy under methoxycarbonylation conditions. The reaction of the complex [PdCl2(1)2] (1a) with pTsOH yields the dimeric complex [PdCl(μ-Cl)(1)]2 (1b), which was characterized by multinuclear NMR spectroscopy and X-ray crystallography. The isolated complex 1b was tested as a precursor in the methoxycarbonylation of styrene, providing poor activity and enantioselectivity. Evidences for the protonation of ligand 1 under catalytic conditions were obtained. When complex [Pd(OTs)(OH2)(R,R-bdpp)][OTs] (2b) was used as the catalyst precursor evidences for both hydride and methoxycarbonylation mechanisms were achieved. Two isomeric forms of [Pd(π-methylbenzyl)(R,R-bdpp)], 2h and 2i, were characterized by multinuclear NMR spectroscopy and DFT calculations. The results obtained by DFT calculations showed that the two most stable isomers contain the ligand bdpp in the “chair” conformation and that both isomers have very similar energies. The hydrido-carbonyl-bridged complex [Pd2(μ-H)(μ-CO)(R,R-bdpp)2][OTs] (2e) was also detected under catalytic conditions. Deuterium labeling experiments using [D4]MeOH and GC-MS analysis of the unconverted substrate and products were performed and the results indicate that the styrene insertion step into the Pd-H bond of the catalyst is irreversible when the phosphetane containing system is used, whereas this step is reversible when the diphosphane precursor is used as the incorporation of deuterium was only observed in the latter case.