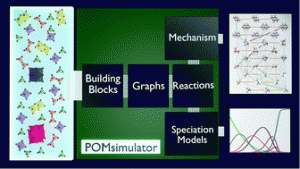
The self-assembly mechanisms of polyoxometalates (POMs) are still a matter of discussion owing to the difficult task of identifying all the chemical species and reactions involved. We present a new computational methodology that identifies the reaction mechanism for the formation of metal-oxide clusters and provides a speciation model from first-principles and in an automated manner. As a first example, we apply our method to the formation of octamolybdate. In our model, we include variables such as pH, temperature and ionic force because they have a determining effect on driving the reaction to a specific product. Making use of graphs, we set up and solved 2.8·105 multi-species chemical equilibrium (MSCE) non-linear equations and found which set of reactions fitted best with the experimental data available. The agreement between computed and experimental speciation diagrams is excellent. Furthermore, we discovered a strong linear dependence between DFT and empirical formation constants, which opens the door for a systematic rescaling.