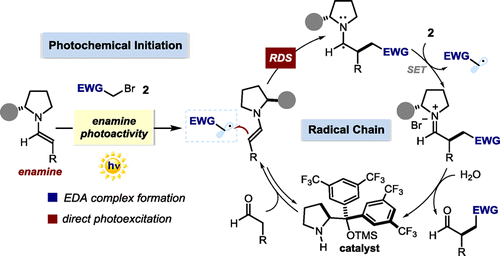
Herein we describe our efforts to elucidate the key mechanistic aspects of the previously reported enantioselective photochemical α-alkylation of aldehydes with electron-poor organic halides. The chemistry exploits the potential of chiral enamines, key organocatalytic intermediates in thermal asymmetric processes, to directly participate in the photoexcitation of substrates either by forming a photoactive electron donor-acceptor (EDA) complex or by directly reaching an electronically excited state upon light absorption. These photochemical mechanisms generate radicals from closed-shell precursors under mild conditions. At the same time, the ground state chiral enamines provide effective stereochemical control over the enantioselective radical trapping process. We use a combination of conventional photophysical investigations, nuclear magnetic resonance (NMR) spectroscopy, and kinetic studies to gain a better understanding of the factors governing these enantioselective photochemical catalytic processes. Measurements of the quantum yield reveal that a radical chain mechanism is operative, while reaction-profile analysis and rate-order assessment indicate the trapping of the carbon-centered radical by the enamine, to form the carbon-carbon bond, as rate-determining. Our kinetic studies unveil the existence of a delicate interplay between the light-triggered initiation step and the radical chain propagation manifold, both mediated by the chiral enamines.