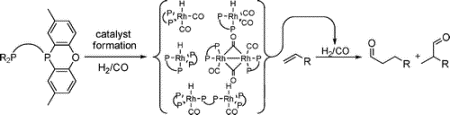
The synthesis of a new series of diphosphine ligands based on 2,7-di-tert-butyl-9,9-dimethylxanthene (1), p-tolyl ether (2), ferrocene (3), and benzene (4) backbones, containing one or two 2,8-dimethylphenoxaphosphine moieties, is reported. The ligands were employed in the rhodium-catalyzed hydroformylation of 1-octene. For all four ligand backbones, introduction of phenoxaphosphine moieties led to an increase in catalytic activity and a decrease in regioselectivity toward the linear aldehyde product. Xanthene-based ligands 1a–1c yielded highly active and regioselective hydroformylation catalysts; ligands containing p-tolyl ether and ferrocene backbones 2a–2c and 3a–3c provided less active and less regioselective catalysts. Catalysts containing benzene-derived ligands 4a and 4b showed a remarkable preference for the formation of the branched aldehyde product. The coordination behavior of ligands 1–4 under hydroformylation conditions was investigated using high-pressure NMR and IR spectroscopy, revealing the distinct steric and electronic properties of the diphenylphosphine and 2,8-dimethylphenoxaphosphine moieties in ligands 1–4. The phosphacyclic moieties proved to be less basic and less sterically demanding toward other ligands in metal complexes than the acyclic diphenylphosphine moieties. For ligands that contain rigid backbones, the lack of conformational freedom in these phosphacyclic moieties does lead to repulsive interactions between the substituents of the two phosphorus donor atoms, resulting in an increase in the bite angle of the ligand. The low catalytic activity of rhodium catalysts modified by benzene-based ligands 4a–4c was attributed to the quantitative formation of HRh(L)2 under hydroformylation conditions.