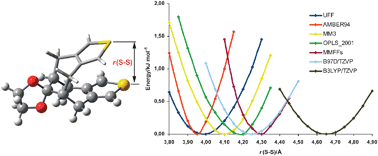
The performance of ONIOM(DFT:MM) methods on the structural description of molecules where intramolecular non-covalent interactions play a critical role is examined systematically and compared with that of full DFT methods, both with and without explicit dispersion corrections. The more detailed study is carried out on dithienobicyclo-[4.4.1]-undeca-3,8-diene-11-one ethylene glycol ketal molecule (CSD entry:RESVAN). Accurate description of the non-covalent interactions between two thiophene rings, measured by the optimal S-S distance (X-ray structure 4.29 Å and estimated experimental gas-phase value 4.19 Å), is accomplished by ONIOM(B3LYP:OPLS2001) [r(S-S) ≈ 4.20 Å] and ONIOM(B3LYP:MMFFs) [r(S-S) ≈ 4.30 Å] calculations, providing results of similar quality to those of dispersion energy corected density functionals such as B97D [r(S-S) = 4.25 Å], B3LYP-D [r(S-S) = 4.25 Å], and TPSS-D [r(S-S) = 4.22 Å], and much more accurate than those of more conventional d. functionals such as B3LYP [r(S-S) = 4.65 Å]. The trends were confirmed by calculations on three other molecules with ONIOM(B3LYP:UFF) and full DFT methods.