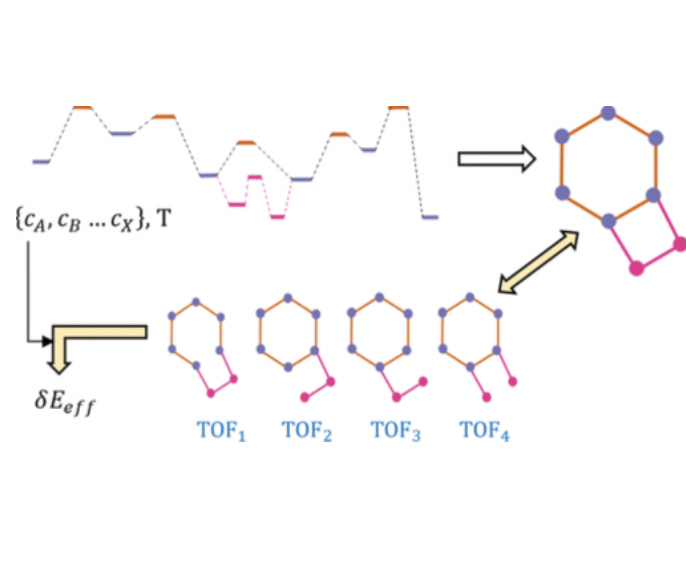
The computational study of catalytic processes allows discovering really intricate and detailed reaction mechanisms that involve many species and transformations. This increasing level of detail can even result detrimental when drawing conclusions from the computed mechanism, as many coexisting reaction pathways can be in close competence. Here, we present a reaction network-based implementation of the energy span model in the form of a computational code, gTOFfee, capable of dealing with any user-specified reaction network. This approach, compared to microkinetic simulations, enables a much easier, simpler, and straightforward analysis of the performance of any catalytic reaction network. In this communication, we will go through the foundations and applicability of the underlying model and will tackle the application to two relevant catalytic systems: homogeneous Co-mediated propene hydroformylation and heterogeneous CO2 hydrogenation over Cu(111).