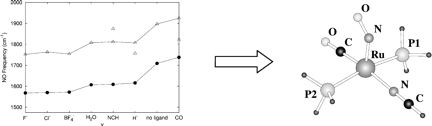
The metal nitrosyl complexes RuX(CO)(NO)L2+ (L ) P(tBu)2Me, X = F–, Cl–, BF4–, H2O, NCH, H–, no ligand, and CO) have been characterized by computing their structures, relative stabilities, and vibrational frequencies through Becke3LYP calculations on a RuX(CO)(NO)– (PH3)2+ model complex. In the case of X = F–, Cl–, BF4–, and H2O, a square pyramidal (SP) geometry with a bent nitrosyl ligand is preferred. In the case of X = NCH, H–, and CO two geometries exist as local minima: a trigonal bipyramid (TBP) with a linear nitrosyl ligand and a square pyramid with a bent nitrosyl ligand. The computed relative stabilities of such complexes cannot clearly identify the ruthenium coordination geometry. Nevertheless, the correlation between the experimental and theoretical îNO stretching frequencies is conclusive in identifying Ru(H)(CO)(NO)L2 and Ru(CO)2(NO)L2+ as TBP structures and Ru(NCMe)– (CO)(NO)L2+ as SP. Three sets of additional calculations were also carried out on a selected system (X = H-). The computational level was increased to CCSD(T), the solvent effect was introduced with a PCM approach, and the real phosphine ligands were considered with a QM/MM ONIOM method.