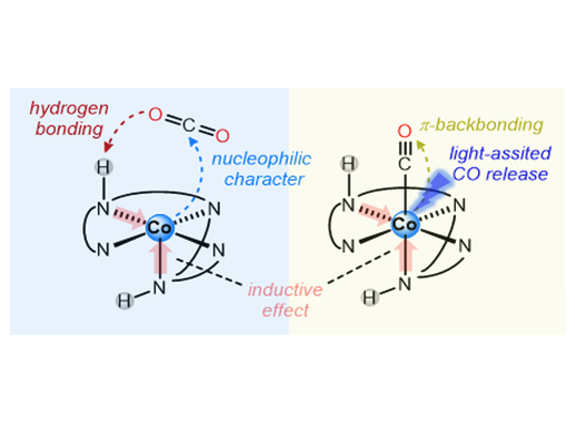
CO2 electroreduction could be improved by applying conceptualized strategies to overcome catalytic bottlenecks. In this regard, we report two new cobalt(II) complexes [Co(Py2R N3)(OTf)](OTf) (CoR , R = H, Me) based on a new C2 -symmetric pentacoordinate chiral ligand that are active on the electrochemical CO 2 reduction to CO. One of the complexes has a N–H group oriented towards the CO2 binding site (CoH ), while the other has a N–Me group with the same orientation (CoMe ), as showed by X-ray diffraction. We have studied the effect of introducing hydrogen bonding sites, i.e. N–H in Co H , as a strategy to stabilize reaction intermediates. The complex bearing coordinating unprotected N–H group (Co H ) displays catalytic CO2 reduction at the CoII/I redox potential (-1.9 V vs. Fc, ca. 40% FYCO). Whereas CoMe shows CO2 reduction at the CoI/0 redox pair. FTIR-SEC and DFT calculations allowed for the identification of a [CoI-CO]+ cation as a catalytic intermediate. The beneficial effect of the N–H group has been attributed to the stabilization of reaction intermediates or transition states and by the larger electron-donating capacity enhancing the nucleophilic character of the CoI intermediate. The study also points to the CO dissociation from the Co(I)-CO resting state intermediate as one of the bottlenecks of the catalytic cycle, which can be overcome with light irradiation, resulting in an increase of the total CO production (-1.9 V, 81% FYCO, 11 TONCO) at the CoII/I potential.