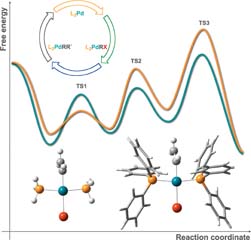
Density functional theory (DFT) and density functional theory/molecular mechanics (DFT/MM) methods are useful tools in modern homogeneous catalysis. Calculation, with its ability to characterize otherwise hardly accessible intermediates and transition states, is a key complement to experiment for the full characterization of the often intricate reaction mechanisms involved in transition metal catalysis. DFT and DFT/MM techniques have been applied to the characterization of full catalytic cycles, as those in cross-coupling; to the systematic analysis of single reaction steps common to several catalytic cycles, such as CH activation; to the elucidation of processes involving different spin states, such as the rebound mechanism for CH activation; to the identification of transient intermediates with key mechanistic roles, such as those in oxygen-evolving complexes; to the analysis of the catalytic keys to polymerization control, as in olefin polymerization; and to reproduction and rationalization of experimentally reported enantioselectvities, as in the case of olefin dihydroxylation. The currently available techniques provide sufficient accuracy to offer chemical insight into the systems involved in experiment, as proved by the growing body of successful applications in the field.