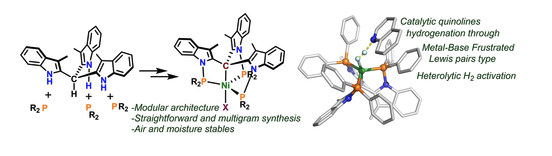
The reactivity of metal complexes can be primarily controlled by their coordinating ligands. Electronic effects and geometric restrictions imposed by ligands, prototypically modify their reactivity, but are not always straightforward accessible. In the course of the study, we explored new C 3 -symmetric organometallic nickel (II) complexes (( P 3 C )Ni )) with the particularity of having a metalated apical carbon trans to a labile coordination site readily available as a catalytic site. The synthetic methodology allowed to tune the electronic properties by introducing substituents on the phosphines. We studied the nature of the C-Ni bond by X-ray absorption spectroscopy (XAS) and DFT, and applied them to the catalytic hydrogenation of quinones with a good chemical scope and group tolerance. 1 H-NMR monitoring, isotopic labelling, and computational studies are consistent with a heterolytic activation of the dihydrogen molecule after forming a s-H 2 adduct trans to the metalated carbon.