The Suero research group has developed new catalytic methods involving conceptually-novel radical carbenoids, carbyne equivalents, and metal-carbynoids and studied their behavior towards fine & feedstock chemicals as well as medically relevant agents. Key to this work has been to design and synthesize tailored reagents that upon photoredox, light or metal activation delivered the novel reactive species. Those reagents have found unique applications in the late-stage functionalization of drug molecules and natural products, that permitted the construction of chiral centers using C–H bonds as functional groups and in cyclopropanations of styrenes. In addition, the group is interested in the development of (i) high-throughput synthetic technologies to enable the synthesis of libraries of bioactive molecules with new chemical space, (ii) new reagents for peptide/protein bioconjugation reactions, and (iii) radiolabelling reactions for the discovery of new radiotracers to impact PET imaging.
New cyclopropanation reactions with radical carbenoids enabled by photoredox catalysis.
The catalytic transfer of a methylene group to alkene molecules for the synthesis of cyclopropane cores is a long-standing challenge in chemical synthesis. At present, classical protocols rely on the use of iodomethylzinc reagents, diazomethane or sulfur ylides, which possess problems with efficiency, availability, and safety. Although reliable control of the stereochemistry is achieved, these processes remained confined to the utilization of isomeric pure alkenes.
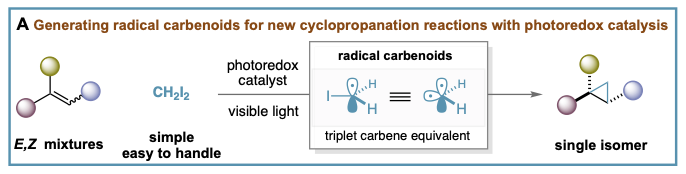
Our group reported the first stereoconvergent cyclopropanation reaction by means of photoredox catalysis using diiodomethane as the methylene source (A). We have demonstrated that the process involves iodomethyl radical (•)CH2-I, a triplet carbene equivalent that we termed radical carbenoid, able to transfer a CH2 group to a wide range of E,Z-alkene mixtures in a stereocontrolled manner. Notable features of this process are its mild conditions, broad functional group compatibility, and excellent selectivity profile. This work resulted in two publications, Angewandte Chemie and European Journal of Organic Chemistry, and was highlighted on the cover of Angewandte.
 After this, the groups of Molander, Charette, Li and Ooi reported complementary methods generating same or analogous carbenoid-like radicals. A short review highlighting the new radical cyclopropanation was published in Synthesis by our group (Invited review by Prof. Paul Knochel).
After this, the groups of Molander, Charette, Li and Ooi reported complementary methods generating same or analogous carbenoid-like radicals. A short review highlighting the new radical cyclopropanation was published in Synthesis by our group (Invited review by Prof. Paul Knochel).
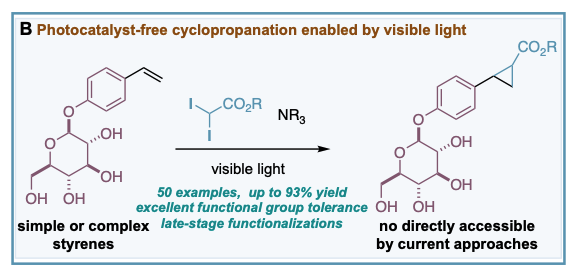
Control experiments carried out during the development of our photoredox-catalyzed cyclopropanations, revealed that excluding [Ru(bpy)3][PF6]2 catalysts in the reaction resulted in a poor yield of the corresponding cyclopropane (18% yield, 18 hours). This interesting result encouraged us to question whether gem-diodomethyl carbonyl reagents – activated alkyl iodides with more absorbance in the visible region – could provide an efficient photocatalyst-free cyclopropanation and deliver valuable carbonyl-substituted cyclopropane rings. We have developed a general photocatalyst-free cyclopropanation of styrenes that use underexploited gem-diodomethyl carbonyl reagents and a simple compact fluorescence lamp (CFL) as visible light source (B). The process generates valuable cyclopropyl carboxylates that can be easily diversified by well-documented methods. Its excellent functional group tolerance and chemoselectivity have enabled access to cyclopropyl carboxylates that are not possible to obtain in a direct manner by classic or current catalytic strategies relying on metal-carbene species (see example below). This work was published in Chemical Science.
 Generating carbyne equivalents with photoredox catalysis
Generating carbyne equivalents with photoredox catalysis
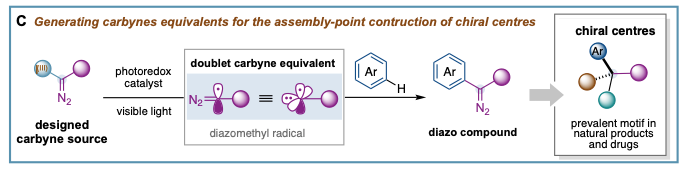
We have pioneered the generation of neutral carbyne equivalents by means of photoredox catalysis (C). This study revealed the underexploited dual carbene/radical behavior and ability of neutral carbynes to form three new bonds; and provided the fundaments to an “assembly-point” functionalization for chiral centre sequencing through diazo compounds. Key to this study was the design of stable carbyne sources decorated with a hypervalent iodine moiety I(III) (I(III) represents − I(III) (Ar)(OTf)) and a diazo functionality (=N2). Catalytic single-electron reduction selectively “unmasked” the I(III) moiety, generating an electrophilic diazomethyl radical (N2=C(•)-R) as direct carbyne equivalent, able to cleave aryl C−H bonds in feedstock chemicals and medically relevant agents.
 This work was published in Nature and highlighted on Nature News & Views, CEN, Chemistry World and mentioned on the cover of Nature. The innate ability of monovalent carbons to form three new bonds suggest that methodologies based on carbynes, may have the ability to streamline chemical synthesis and to reach previously unattainable chemical space in the pharmaceutical sector (among others). In fact, this work has recently been highlighted by medicinal chemists from Merck in a review of Science magazine (Science 363, (2019) eaat0805) as the latest of a series of very diverse, practical, and potentially impactful uses of photoredox techniques to assemble libraries of drug-like scaffolds for screening, and also in Nature Reviews Drug Discovery journal (Nat. Rev. Drug Discov. 17, 709 (2018)) by medicinal chemists from Astra Zeneca and GSK.
This work was published in Nature and highlighted on Nature News & Views, CEN, Chemistry World and mentioned on the cover of Nature. The innate ability of monovalent carbons to form three new bonds suggest that methodologies based on carbynes, may have the ability to streamline chemical synthesis and to reach previously unattainable chemical space in the pharmaceutical sector (among others). In fact, this work has recently been highlighted by medicinal chemists from Merck in a review of Science magazine (Science 363, (2019) eaat0805) as the latest of a series of very diverse, practical, and potentially impactful uses of photoredox techniques to assemble libraries of drug-like scaffolds for screening, and also in Nature Reviews Drug Discovery journal (Nat. Rev. Drug Discov. 17, 709 (2018)) by medicinal chemists from Astra Zeneca and GSK.
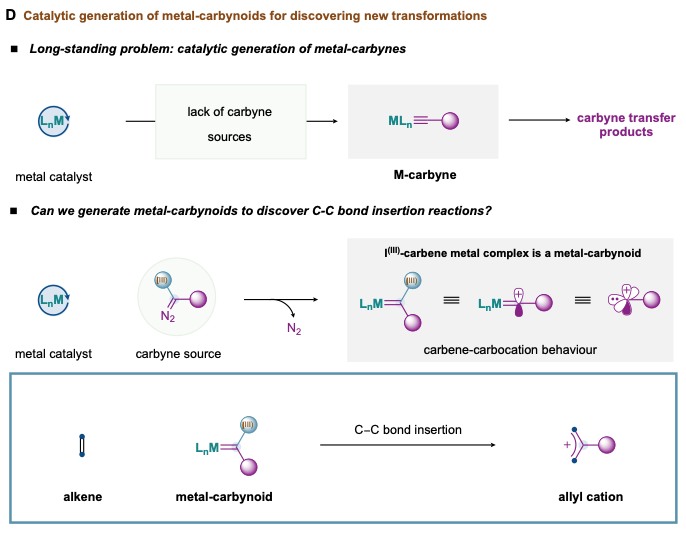
Catalytic cleavage of carbon–carbon double bonds with Rh-carbynoids
For more than half a century, the discovery of new metal-carbon bond-forming strategies has been cornerstone in the development of transition-metal catalysis. The catalytic formation of organometallic intermediates with metal-carbon single/double bond – such as metal-L (L = alkyl, alkenyl, alkynyl, aryl), metal-carbene (metal=L) – is commonly used in reaction discovery as well as in the synthesis of natural products, pharmaceuticals, and functional materials. However, while metal-carbynes – the organometallic species with a metal-carbon triple bond (metal≡L) -, have been key players as catalysts in alkyne metathesis, their general application in catalytic carbyne transfer processes to organic matter has been largely unexplored; mainly due to the lack of suitable carbyne sources that enable the catalytic monovalent ligand transfer. To circumvent this problem, we sought to develop a platform based on the catalytic generation of equivalent forms of metal-carbynes or metal-carbynoids with dual carbene/carbocation behaviour able to uncover a new catalytic alkene activation (D). Recently, we have discovered a catalytic strategy that generates rhodium-carbynoids by selective diazo activation of designed carbyne sources with rhodium paddlewheel complexes. We found that our equivalents provoke C-C bond scission by inserting a cationic monovalent carbon unit between both sp2-hybridized carbons. This skeletal remodeling process accesses synthetically useful allyl cation intermediates that conduct to valuable allylic building blocks upon nucleophile attack. Our results rely on the formation of cyclopropyl-I(III)intermediates able to undergo electrocyclic ring-opening, following the Woodward–Hoffmann–DePuy rules. Furthermore, the insertion of a monovalent carbon unit in C-C bonds underscores an opportunity in skeletal editing that will be relevant to reach previously unattainable chemical space in drug discovery. This work was published in the Journal of the American Chemical Society in September 2019 and has been seen over 12000 times on the JACS ACS web, highlighted in Chemistry Views and Chemistry World, and was one of the most read articles in September 2019 and was among the Top 20 most downloaded and most-read articles within the last 12 months of publication.